健康知识库
劳吉尔-亨齐克尔综合征
编辑:ddayh.cn
- 上一篇:寄生虫妄想症
- 下一篇:进行性色素性紫癜性皮病
- 200痤疮(青春痘)的防治
- 200淋病
- 200睡眠是大自然赋予的神奇恢复剂,
- 200立夏|夏季阳气盛于外,忌扼杀阳
- 200治疗湿疹的秘密被揭开了,你知道
- 200霉菌性阴道炎 是孕期常见疾病,
- 200关爱白癜风患者,从我做起!
- 200血管瘤
- 200寒冷季节如何防治冻疮
- 200孕妇应该怎样预防荨麻疹
- 200什么是皮肤屏障?
- 200Hpv感染的种种问题
- 200欢迎大家前来咨询
- 200二期梅毒的主要症状有哪些
- 200带状疱疹
- 200为什么防晒要从春天做起?及防晒
- 200梅毒经过了正规治疗,可滴度却不
- 200老年斑和老年疣的区别
- 200最近几天频繁荨麻疹怎么办
- 200痤疮是由什么引起的
劳吉尔-亨齐克尔综合征(Laugier-Hunziker Syndrome)
本病是一种唇、口腔粘膜和指(趾)甲获得性色素沉着性疾病,由Laugier-Hunziker于1970年首先报道。此后报道的病例多来自欧洲,其他地区则较少见。我国马东来等于2006年报道8例,说明本病在我国可能不少见。本病病因不明,有作者认为可能是由于某种未知的刺激,使基底层黑素细胞的功能发生改变,合成黑素小体的数量增加所致。本病多散发,仅有一个家族发病的报道。
【临床表现】
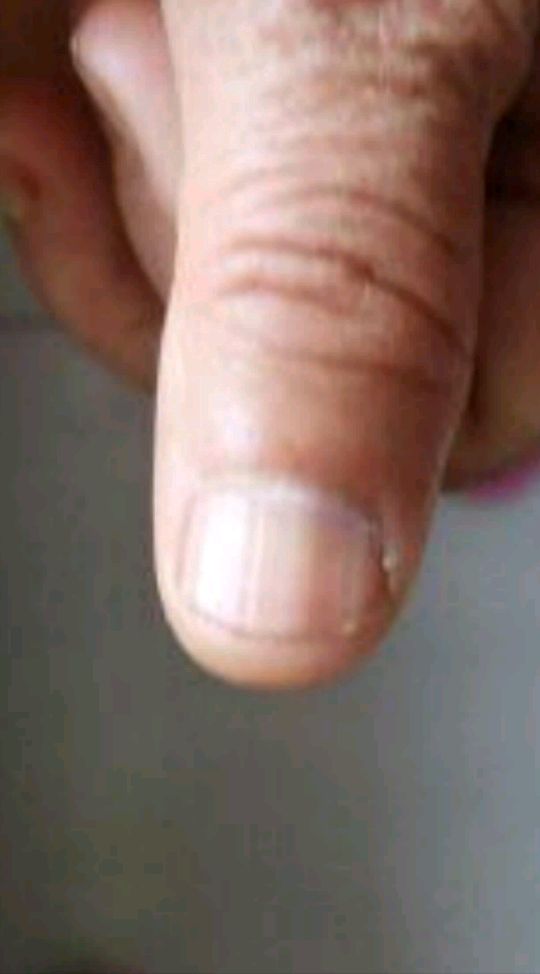
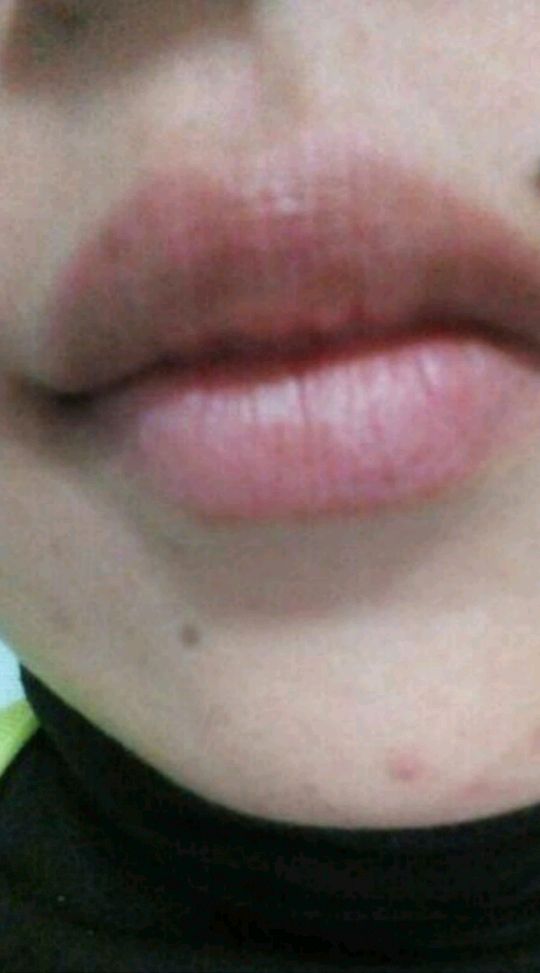
男女发病率相近,皮损一般在30-50岁才出现。好发于唇部、颊粘膜、硬腭和指(趾)甲,但偶可累及口周、牙龈、舌、喉、软腭、腭舌弓、口腔底部、食管、颈部、胸部、手指、足趾、掌跖、结膜、阴茎和肛周等部位。皮损为圆形、卵圆形或不规则形的色素沉着斑。可单发、群集或融合成片,偶尔克成线状,表面光滑,呈灰色、褐色或蓝黑色,边界清楚或模糊。一般无自觉症状。60%的患者有甲的色素沉着,主要有下列四种形式: ①单一的色素性纵条,1~2mm宽;②甲板两侧各有一条2~3mm宽的色素带;③半侧甲有均一的色素沉着;④全甲色素沉着。同一患者的不同指(趾)甲可同时存在上述四种色素沉着斑,但一般不伴有甲营养不良。少数患者Hutchinson征可阳性(线状黑甲末端的甲皱出现黑素沉着),但不会发展成黑素瘤。本病不伴有结肠息肉,无腹痛、腹泻、呕吐和便血等症状。本病病程慢性,皮损常进行性加重,仅有1例患者有皮损自行消退的报道。
【组织病理】
无特异性,主要表现为表皮基地细胞内黑色素增多,但黑素细胞的数量和形态正常,基地细胞膜完整。真皮浅层色素失禁,可见数量不等的噬黑色细胞。
【诊断及鉴别诊断】
本病主要应与Peutz-Jeghers 综合征鉴别,后者为常染色体显性遗传,可有STK II/LKB I位点的基因突变。一般在出生时或出生后不久发生,皮损多位于口腔、鼻子等腔口周围,组织病理为雀斑样痣表现。多伴肠息肉。
【治疗】
本病色素沉着不会恶变为黑素瘤,因此一般不需治疗。为美容目的,色素沉着斑可用Q开关紫翠宝石激光治疗。
(图1 右小趾甲板全甲色素沉着)
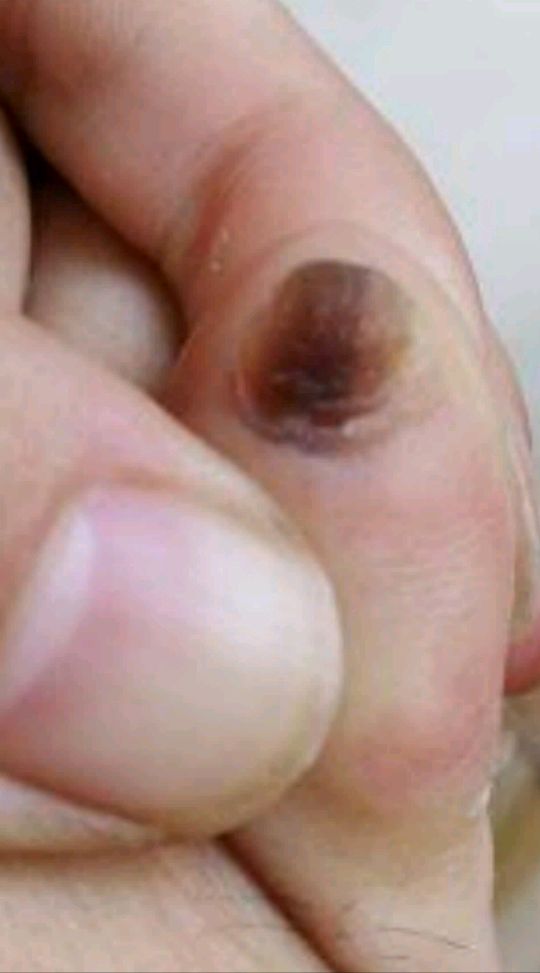
(图2 右足2趾甲板色素带)
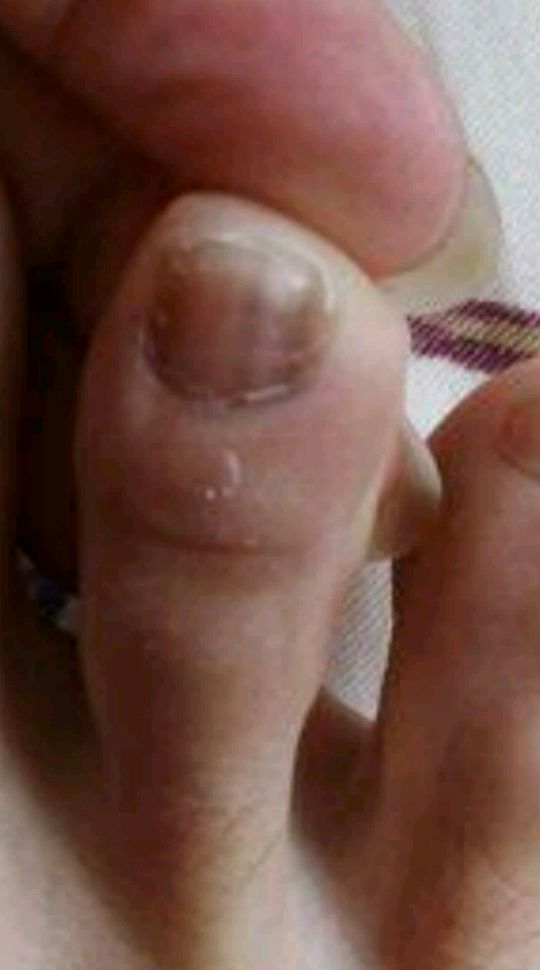
(图3 左手拇指甲板多条色素带)
(图4 上唇粘膜色素沉着斑)
热门皮肤科